
CMT2A ASPETTI CLINICI
CMT 2A2: NEUROPATIA ASSOCIATA A MUTAZIONI DEL GENE MITOFUSINA 2
(MFN 2, Cromosoma 1p36.22)
Epidemiologia
Ne sono affetti il 19-23% (in uno studio pubblicato dalla rivista scientifica Brain nel 2006 fino al 33%) dei soggetti con neuropatia assonale dominante tipo CMT2 (neuropatia assonale ereditaria a trasmissione autosomica dominante), rappresentandone pertanto il sottotipo più frequente.
La penetranza è variabile (all’interno della stessa famiglia più soggetti affetti possono presentare quadri differenti in caratteristiche e gravità).
Ereditarietà
La mutazione nel gene della mitofusina 2 responsabile di neuropatia assonale è prevalentemente trasmessa con carattere autosomico dominante, cioè un genitore portatore di una mutazione a carico del gene MFN2 può trasmetterla ad un figlio, che pertanto sarà affetto da malattia, con una probabilità del 50% .
In casi più rari la trasmissione genetica è recessiva omozigote, cioè entrambi i genitori hanno una mutazione su gene MFN2. La probabilità che entrambe le mutazioni siano trasmesse ad un figlio e che pertanto esso sia malato è del 25%. Vi è una probabilità del 50% che un figlio sia portatore di una sola mutazione e del 25% che sia sano. I genitori o i figli portatori di una sola mutazione sono asintomatici o presentano una lieve neuropatia.
Vi sono inoltre mutazioni composte in eterozigosi in trans associate ad un pattern di ereditarietà di tipo autosomico recessivo. Queste ultime determinano spesso un fenotipo ad esordio precoce più severo (Nicholson GA et al, 2008; Vallat JM et al., 2008; JM Polke et al., 2011).
Numerose mutazioni sono invece de novo, senza alcun caso precedente in famiglia
Esordio
L’età di presentazione è variabile, con quadri clinici che esordiscono nell’infanzia (2-3 anni) fino all’ età adulta (quinta decade). Generalmente i casi ad esordio molto precoce sono più gravi
Quadro clinico
Simile a quello ad altre neuropatie ereditarie tipo CMT2 (CMT2A1, CMT2E e CMT2F) nelle quali sono coinvolti altri geni.
In generale la manifestazione principale è una neuropatia ereditaria sensitivo-motoria a lenta progressione, che coinvolge dapprima gli arti inferiori, quindi gli arti superiori, con conseguente difficoltà di deambulazione e debolezza del movimento e dell’utilizzo delle mani.
Il quadro clinico può variare da casi ad esordio precoce con severa debolezza a casi asintomatici (fino al 25% di portatori di mutazioni del gene MTF 2presentano solo segni subclinici).
Lo spettro delle manifestazioni comprende i seguenti ambiti :
> Polineuropatia
- debolezza distale (piedi e mani), soprattutto alle gambe piuttosto che alle braccia, ciò determina problemi nel cammino e conduce in alcuni casi alla perdita dello stesso.
- ipotono e diminuzione/assenza dei riflessi osteotendinei; facile affaticabilità
- disturbi delle sensibilità
> Atrofia ottica, nei casi più gravi
> Dolori, crampi
> Tremori
> Perdita dell’udito
> Macrocefalia
> Ritardo psicomotorio
> Piede cavo, scoliosi
La presenza di alcune di tali caratteristiche è correlabile a tipi specifici di mutazione del gene delle mitofusina (ad oggi sono note più di 80 mutazioni responsabili della patologia).
Nel complesso è possibile distinguere sul piano clinico:
- una forma ad esordio precoce (prima dei 10 anni), caratterizzata da un fenotipo severo, decorso rapido con perdita di autonomia nella deambulazione usualmente entro il ventesimo anno di vita, condizionante pertanto necessità precoce di ricorso ad ausili. Dal punto di vista neurofisiologico questo sottotipo si associa a severe riduzioni in ampiezza del potenziale d’azione muscolare composto (CMAP), a tal punto da non essere registrabile. Questa forma è stata riportata essere associata alle seguenti mutazioni: L92P, R94W, R94Q,T105M, R364W (Zucher et al., 2004; Chung et al., Brain 2006)
- una forma ad esordio tardivo (dopo i 10 anni), caratterizzata da un fenotipo meno severo e da un decorso meno aggressivo, con studi di conduzione neurofisiologici che evidenziano un CMAP nella norma o solo lievemente ridotto. E’ stata osservata in associazione alle seguenti mutazioni nel gene della MFN2: M367T, H165R.
Varianti cliniche della forma classica CMT2A2
- HMSN V/CMT V + spasticità agli arti inferiori (variante con mielopatia)
Trasmissione autosomica dominante con esordio a partire dalla seconda decade di vita. Quadro clinico di CMT2 classica (debolezza agli arti inferiori) con segni piramidali (riflessi osteotendinei vivaci, risposta cutanea plantare in estensione ed in alcuni casi spasticità). A ciò si aggiungono disturbi delle sensibilità e dolori. La progressione è lenta con una modesta disabilità nella maggior parte dei pazienti - HMSN VI /CMTVI+ atrofia ottica
Trasmissione autosomica dominante o sporadica con esordio in età infantile, spesso associata a fenotipi più severi e a mutazioni de novo. Il quadro clinico si caratterizza, accanto alla presenza di una severa neuropatia motoria compatibile con il quadro di CMT, per la comparsa di atrofia ottica con un esordio variabile tra i 5 e i 50 anni, progressiva nei bambini, più lenta negli adulti. Viene descritta una perdita subacuta dell’acuità visiva, con scotomi centro cecali, deficit nella visione dei colori e disco ottico pallido all’esame del fundus oculi, che talvolta nel corso degli anni può andare incontro a vari gradi di regressione con recupero parziale del visus (come osservato in alcuni casi di neuropatia ottica ereditaria di Leber). - CMT + severa encefalopatia
Riscontro alle indagini di neuroimaging di iperintensità di segnale a livello dei peduncoli cerebrali, della sostanza bianca periventricolare e dei centri semiovali, osservabili sia nelle forme ad esordio tardivo (KW Chung et al., Brain 2006) sia nelle forme ad esordio precoce, talvolta associate a secondaria macrocefalia (mutazione R104W) e ad alterazioni microstutturali tipo astrocitosi, aumento della densità neuroassonale e demielinizzazione evidenziata alla SPECT cerebrale (Brockmann et al., Journal of Neurology 2008).Esistono inoltre delle presentazioni cliniche simil “sclerosi multipla” associate a mutazioni nel gene della MFN 2 caratterizzate dalla contemporanea presenza di atrofia ottica progressiva dapprima mono poi bilaterale con alterazione di segnale alla RMN encefalo e solo moderati segni clinici di neuropatia assonale, spesso tra l’altro a comparsa tardiva (mutazione Leu146Phe, Christopher J Klein et al., Archives of Neurology 2011).
- Miopatia mitocondriale con delezioni multiple del DNA mitocondriale da mutazione nel gene MFN 2, ad esordio in età adulta, preceduta da atrofia ottica esordita nell’infanzia e successivo sviluppo di neuropatia assonale (mutazione D210V, Cecile Rouzier et al., Brain 2012).
- Neuropatia recessiva assonale (AR-CMT2)
Esordio infantile, forma più severa con decorso progressivo fino alla perdita del cammino. Può essere presente atrofia ottica o scoliosi All’esame elettrofisiologico CMAP SAP sono assenti o ridotti in ampiezza e le velocità di conduzione nervosa sono normali
Altre varianti
Forma severa, esordio infantile (2-3 anni), progressione veloce con una debolezza diffusa e perdita delle sensibilità fino alla perdita del cammino.
Dal punto di vista genetico i soggetti affetti possono manifestare mutazioni in omozigosi (stessa mutazione su entrambi gli alleli) o in eterozigosi composta (due mutazioni differenti sui due alleli). I genitori sono asintomatici o lievemente sintomatici (lieve neuropatia).
Forma caratterizzata da una compromissione cognitiva con anomalie cerebrali in associazione alla neuropatia assonale.
Diagnosi
Esame neurofisiologico
Attraverso lo studio elettroneuro/miografico (ENG/EMG) si studiano le velocità di conduzione nervosa sensitiva e motoria (VDC) che sono lievemente ridotte nelle neuropatie assonali quali la CMT2, e l’ampiezza dei potenziali d’azione motori e sensitivi (CMAP e SAP) che invece sono significativamente ridotti. Tali esiti indirizzano verso una neuropatia assonale.
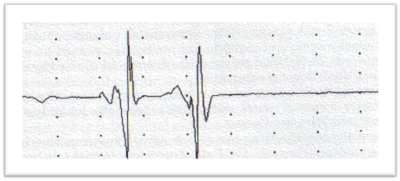
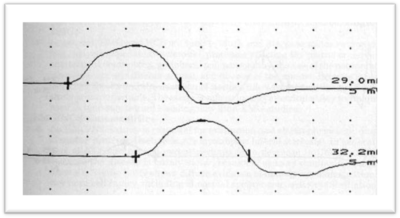
Esame genetico
Se i dati clinici e neurofisiologici indirizzano verso una forma di CMT2A2 si procede all’analisi genetica con in sequenziamento del gene della Mitofusina.
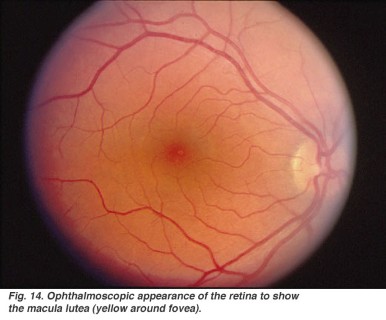
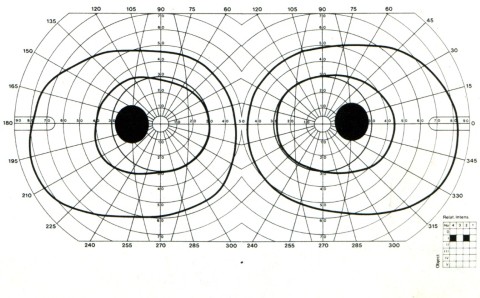
Può risultare utile seguire inoltre una RMN encefalo nel caso siano presenti sintomi riferibili al Sistema Nervoso Centrale, una visita oculistica con esame del Fundus Oculi e studio del Campo Visivo in caso di presenza di calo del visus e biopsia del nervo surale nel caso di diagnosi dubbia e di necessità di acquisire informazioni aggiuntive morfostrutturali o immunoistochimiche a carico del Sistema Nervoso Periferico.
Trattamento
Non essendo ancora disponibile una cura medica risolutiva, l’unica terapia in grado di migliorare le prestazioni funzionali (es. deambulazione, prensione) dei soggetti affetti da CMT è il trattamento riabilitativo neuromotorio.
Elementi fondamentali della riabilitazione della CMT sono le calzature (normali modificate dal tecnico ortopedico oppure su misura), i plantari e i tutori per stabilizzare la caviglia e impedire “la caduta” del piede durante il cammino.
La fisioterapia è importante per prevenire le deformità articolari e per migliorare le prestazioni funzionali dopo l’adozione di calzature ed ortesi appropriate.
Il ricorso alla chirurgia ortopedica potrebbe essere utile sia per prevenire o correggere le deformità articolari sia per stabilizzare le articolazioni non più sostenute dalla muscolatura.
Un sostegno psicologico è utile per permettere un percorso di aiuto e accettazione della malattia
